

Die von Hippel-Lindau-Erkrankung ist eine genetische Erkrankung. Die ursächlichen Mutationen im VHL-Gen können von den Eltern vererbt oder auch spontan aufgetreten sein. Ungefähr jeder 36.000ste Mensch ist Träger einer VHL-Erbanlage und somit Betroffener.
Die VHL-Erkrankung tritt typischerweise in Familien gehäuft auf. Sie folgt dabei einem sog. autosomal-dominanten Erbgang. Das bedeutet, dass beide Geschlechter betroffen sein können und keine Generation übersprungen wird. Die Wahrscheinlichkeit der Weitervererbung an ein Kind liegt bei 50 Prozent. Die Erkrankung tritt in der Regel bei den Betroffenen auch klinisch in Erscheinung. Die Ausprägung ist jedoch stark variabel.
Schon bei Geburt lässt sich die Mutation nachweisen. Tumoren treten aber erst mit zunehmendem Alter auf und haben in der Regel frühestens ab dem 6. Lebensjahr Bedeutung. Die meisten Tumoren verursachen zwischen dem 15. und 35. Lebensjahr Krankheitszeichen (Symptome). Die Ausprägung der Erkrankung ist bei Frauen und bei Männern ohne klinisch relevanten Unterschied. Die Altersverteilung sieht wie folgt aus:
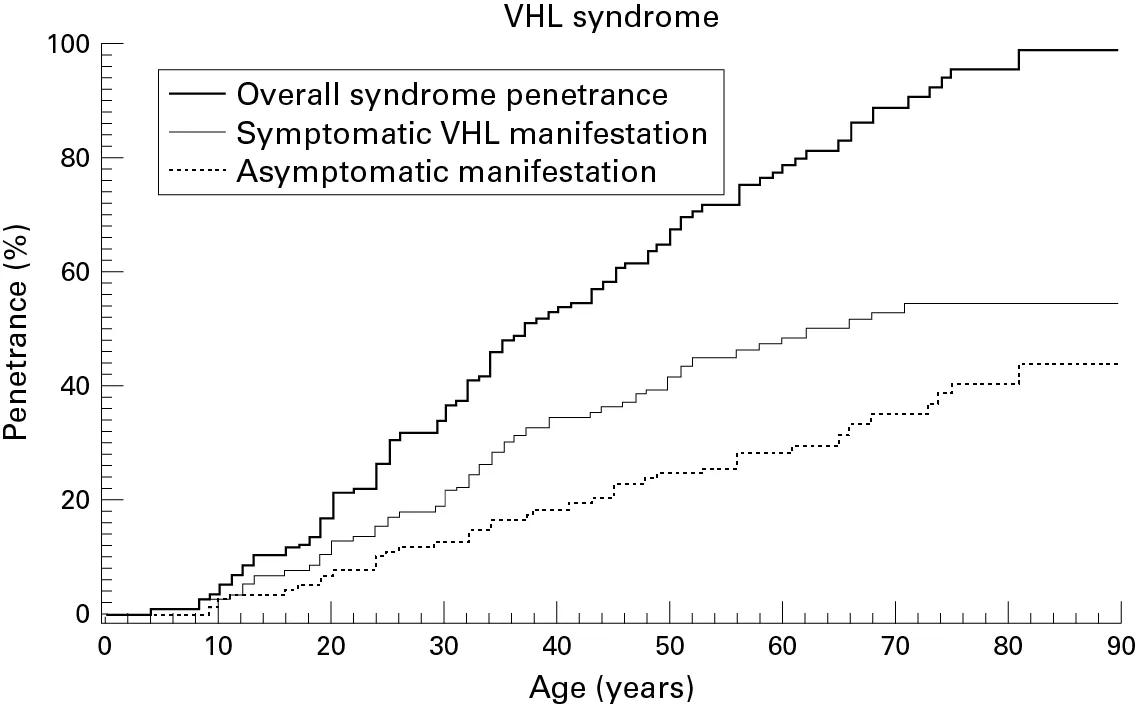
Abbildung Altersverteilung I:
Altersverteilung aller VHL-Veränderungen, sowie der symptomatischen und asymptomatischen Verändeurngen bei 337 VHL Patienten mit der Mutation c505 T/C - Deutschlands häufigster Mutation. Die Kurven stellen eine sog. kumulierte Altersverteilung dar. Man kann entnehmen, mit welcher Wahrscheinlichkeit in Prozent bei einem bestimmten Alter Veränderungen gesehen wurden.
Reproduced from Bender BU, Eng C, Olschewski M, et al; VHL c.505 T>C mutation confers a high age related penetrance but no increased overall mortality;
Journal of Medical Genetics 2001;38:508-514; 2022 with permission from BMJ Publishing Group Ltd.
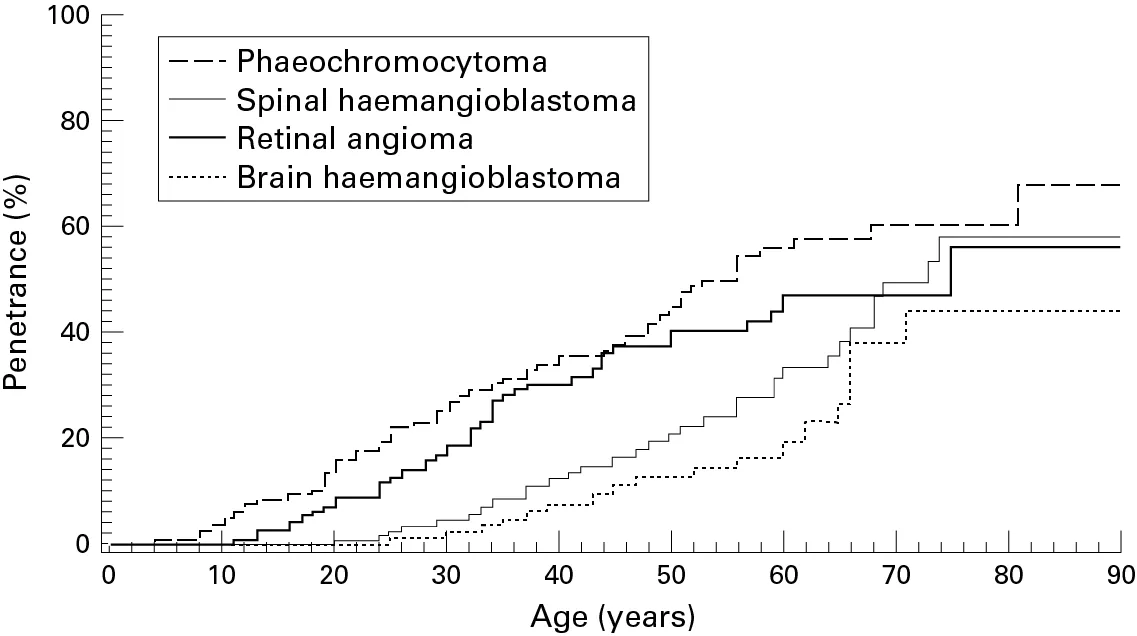
Abbildung Altersverteilung II:
Altersverteilung der Veränderungen von Nebennieren (Phaechromocytoma), Rückenmark (Spinal haemangioblastoma), Augen (Retinal angioma) und Gehirn (Brain haemangioblastoma) bei 337 Patienten mit von-Hippel-Lindau-Erkrankung. Die Kurven stellen eine sog. kumulierte Altersverteilung dar. Man kann entnehmen, mit welcher Wahrscheinlichkeit in Prozent bei einem bestimmten Alter Veränderungen gesehen wurden.
Reproduced from Bender BU, Eng C, Olschewski M, et al; VHL c.505 T>C mutation confers a high age related penetrance but no increased overall mortality;
Journal of Medical Genetics 2001;38:508-514; 2022 with permission from BMJ Publishing Group Ltd.
Die VHL-Erkrankung hat in jeder Familie einen Anfang, d. h. eine Person, bei der sie zum ersten Mal auftritt. Häufig bleibt unklar, wer diese erste Person ist bzw. war. Bei dieser Person ist die Mutation zum ersten Mal vorhanden; solche Mutationen nennt man folglich auch Neu- (oder Spontan-)mutationen. Auch heute werden immer wieder Neumutationen beobachtet. Bei diesen Patienten wird bedauerlicherweise nicht selten die VHL-Erkrankung recht spät erkannt. Es besteht somit die besondere Herausforderung, bei erstmaligem Auftreten der VHL-typischen Tumoren zu prüfen, ob sie Ausdruck der VHL-Erkrankung sind.
Das VHL-Gen wurde bereits 1988 durch molekulargenetische Untersuchungen an Blutproben Angehöriger vieler betroffener VHL-Familien lokalisiert. Sein Genort beherbergt die biologische Information, d. h. die Bauanleitung des VHL-Tumorsuppressors.
Weitere Informationen finden Sie in unsere PKB (Patientenorientierten Krankheitsbeschreibung) sowie in unseren anderen Publikationen.